Comparing TR calls across different genotypers
Tools used: mergeSTR, compareSTR
This vignette shows how to use mergeSTR to merge VCFs from multiple samples into a single VCF, and compareSTR to compare VCF files generated by different genotypers (HipSTR and ExpansionHunter) using the same set of reference TRs. In this example, we use VCF files available at https://github.com/gymrek-lab/TRTools/tree/master/example-files:
NA12878_chr21_eh.sorted.vcf.gz,NA12891_chr21_eh.sorted.vcf.gz, andNA12892_chr21_eh.sorted.vcf.gzgenerated using ExpansionHunter on three separate samplestrio_chr21_hipstr.sorted.vcf.gzgenerated using HipSTR run jointly on all three samples.
Both VCFs were generated using the same reference set of TRs to make them comparable. We’ll also need the file hg19.fa.fai for adding the appropriate header lines to the ExpansionHunter VCF files. (You can generate this file from a reference genome fasta file using samtools faidx).
First, we’ll want to merge the three samples called separately by ExpansionHunter into a single VCF file using mergeSTR. We’ll need to do one VCF cleanup using bcftools reheader to make sure the appropriate contig lines are present in the VCF file:
# Add contig lines to the headers
bcftools reheader -f hg19.fa.fai -o NA12878_chr21_eh.reheader.vcf.gz NA12878_chr21_eh.sorted.vcf.gz
bcftools reheader -f hg19.fa.fai -o NA12891_chr21_eh.reheader.vcf.gz NA12891_chr21_eh.sorted.vcf.gz
bcftools reheader -f hg19.fa.fai -o NA12892_chr21_eh.reheader.vcf.gz NA12892_chr21_eh.sorted.vcf.gz
tabix -p vcf NA12878_chr21_eh.reheader.vcf.gz
tabix -p vcf NA12891_chr21_eh.reheader.vcf.gz
tabix -p vcf NA12892_chr21_eh.reheader.vcf.gz
# Merge all the EH samples to one VCF
mergeSTR --vcfs NA12878_chr21_eh.reheader.vcf.gz,NA12891_chr21_eh.reheader.vcf.gz,NA12892_chr21_eh.reheader.vcf.gz \
--out trio_chr21_eh
# Bgzip and index the output VCF to get ready for compareSTR
bgzip trio_chr21_eh.vcf
tabix -p vcf trio_chr21_eh.vcf.gz
Now, we have a file from each tool (trio_chr21_hipstr.sorted.vcf.gz and trio_chr21_eh.vcf.gz) containing genotypes for all three samples. We can use compareSTR to compare these:
compareSTR --vcf1 trio_chr21_hipstr.sorted.vcf.gz --vcf2 trio_chr21_eh.vcf.gz \
--vcftype1 hipstr --vcftype2 eh --out hipstr_vs_eh
This command tells compareSTR to compare these two VCFs. By specifying --vcftype1 and --vcftype2 we are telling it what format the two VCFs are in. Note, TRTools will try to automatically infer this anyway, and make sure what we infer matches what was specified.
This should output several text files:
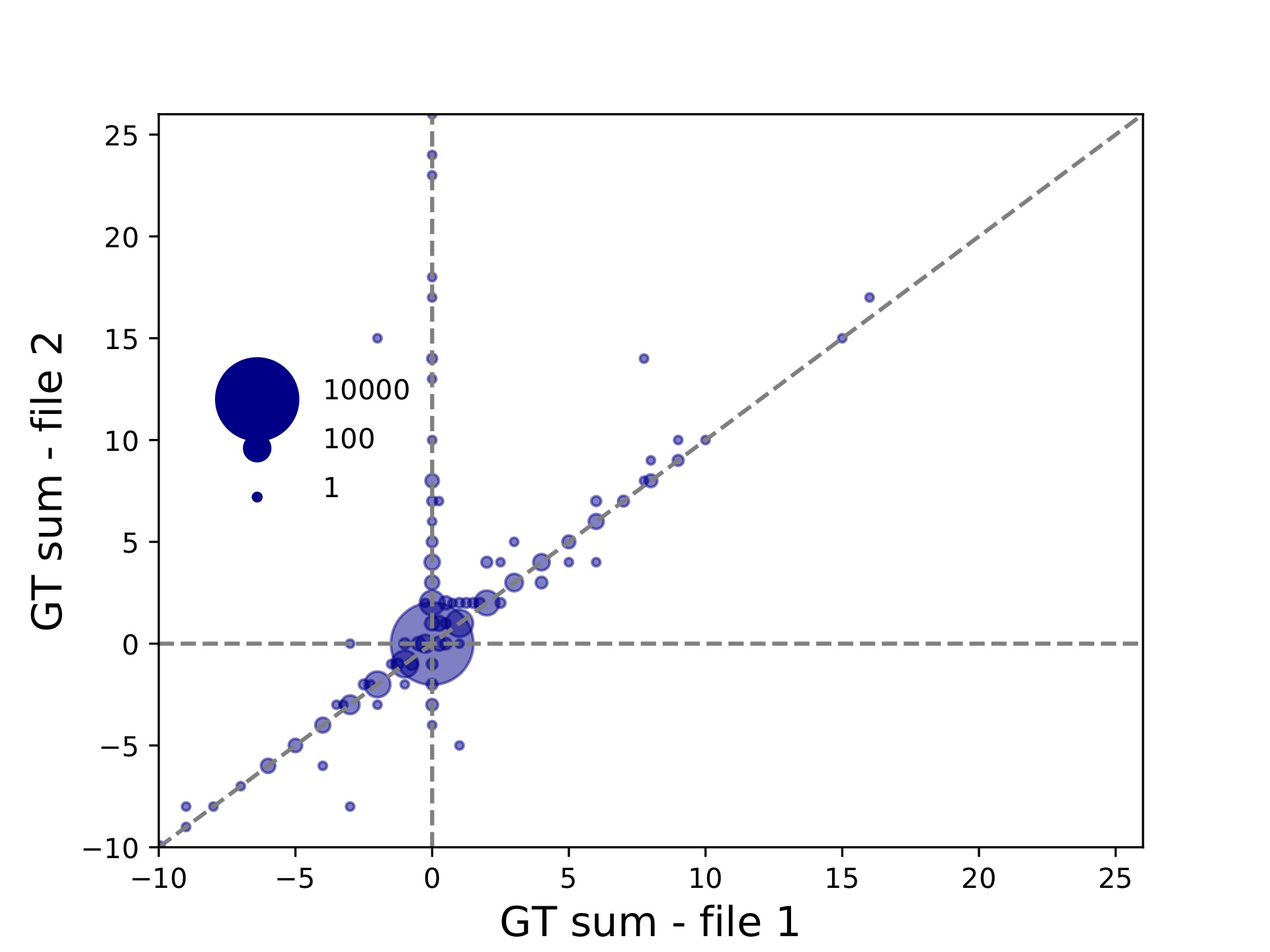
hipstr_vs_eh-overall.tabwill give overall concordance info:period concordance-seq concordance-len r2 numcalls ALL 0.9533784988411481 0.9741486896059903 0.734005622224775 11218
Since we did not specify the --period option, we are considering “ALL” repeat units. This file shows the overall percent of matching calls based on allele sequences (concordance-seq) and allele lengths (concordance-len), the squared Pearson between genotypes (sum of allele lengths) (r2), and the number of calls compared (numcalls).
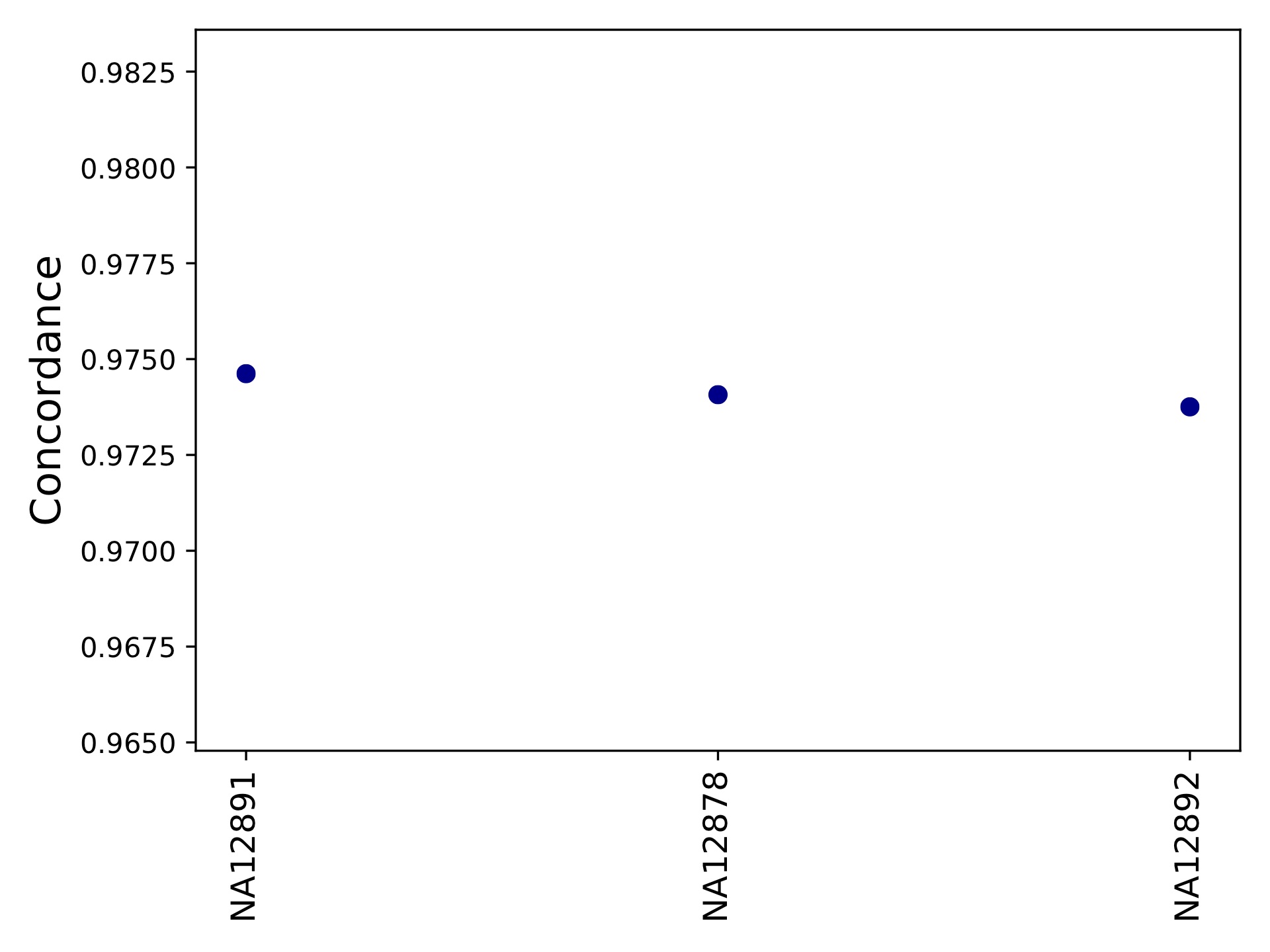
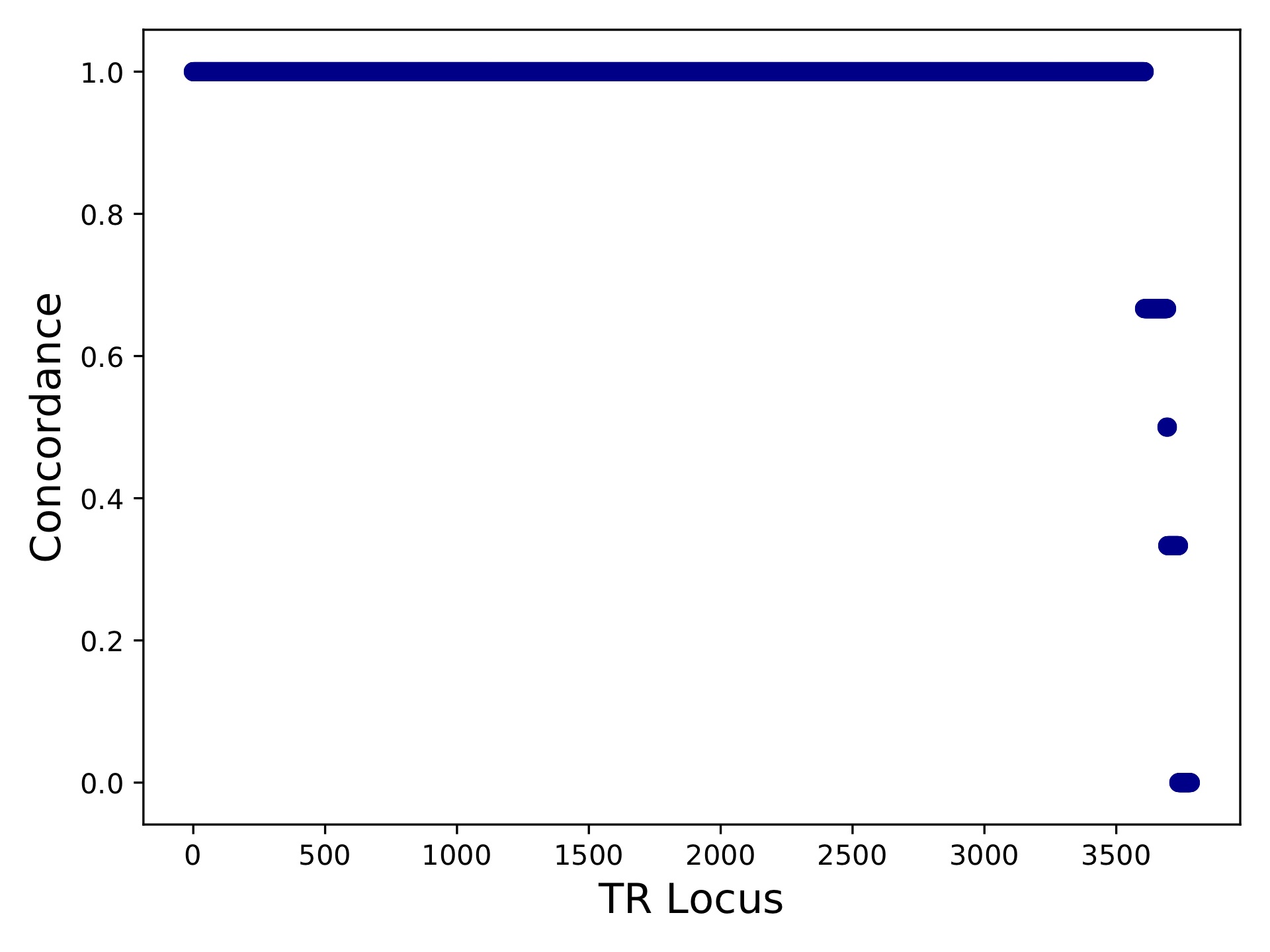
hipstr_vs_eh-callcompare.tabgives a call by call comparison.hipstr_vs_eh-locuscompare.tabgives per-locus comparison statistics.hipstr_vs_eh-samplecompare.tabgives per-sample comparison statistics::sample metric-conc-seq metric-conc-len numcalls NA12891 0.9540475554368154 0.9746192893401016 3743 NA12878 0.9526864474739375 0.9740711039828923 3741 NA12892 0.953401178361007 0.9737546866630958 3734
mergeSTR will also output several plots to visualize the comparison results:
hipstr_vs_eh-samplecompare.pdf
hipstr_vs_eh-locuscompare.pdf
hipstr_vs_eh-bubble-periodALL.pdf